
RNA-seq解析|受託解析
RNAseq解析とは、次世代シーケンサー(NGS)を用いて転写物の塩基配列を決定する方法です。この配列をリファレンスゲノムへアライメントし、転写産物毎の発現量を測定し、通常はサンプル間の発現量の比較解析を行います。スプライスバリアント毎の発現解析、融合遺伝子検出、変異解析、新たな転写産物の予測を行うことができます。
リファレンスゲノムのない生物の場合でも、配列をアセンブルして転写産物モデルを構築し、アライメントさせて解析することができます。
サービス内容
【WET】RNAシーケンス| ○ ライブラリ調製 |
| ○ Illumina社 NovaSeq™ X Plus / MGI社 DNBSEQ™ によるシーケンシング(PolyA、150PE) |
| ○ 発現定量解析:マッピング、発現定量、階層的クラスタリング (ヒートマップ描画)、 主成分分析 |
| ○ 二群間の発現量比較解析(サンプル数によって組み合わせ数を決定):Volcano plot |
| ○ エンリッチメント解析:Gene Ontology解析、Reactomeパスウェイ解析 |
| ○ 解析レポート、結果データ一式の納品 |
サンプル要件 / 注意事項
サンプルタイプ:total RNA
| 総量(ng) | 液量(µL) | 濃度(ng/µL) | RIN値 | 純度 |
|---|---|---|---|---|
| 200以上 | 10以上 | 20以上 | 4.0以上 with flat base line |
OD260/280 = 1.8-2.2 OD260/230 ≥ 1.8 no degradation, no contamination |
- ※ サンプルQCでRNA総量またはRIN値が非常に低い場合は、再提出をお願いすることがございます
(再提出1回まで無償対応) - ※ シーケンシングは日本(NovaSeq™:真核生物のみ)またはシンガポール・中国にて実施します
- ※ 対応生物種:ヒト・マウス・ラット
その他の生物種はご相談ください - ※ RNA抽出からご依頼時のサンプル要件: 分離細胞1.0×105 細胞以上、組織切片10-30 mg
- ※ DRY(データ解析)のみの場合、FASTQファイル(生データ)から受付いたします
データ解析内容
オプション
研究目的に応じて、生物種対応や深掘り解析、高次解析の追加なども対応いたします。
| 分類 | オプション |
|---|---|
| 【WET】 RNAシーケンス |
データ増量(+1Gb) |
| ストランド特異性 | |
| 9Gb(+5Gb)+ストランド特異性 | |
| 【DRY】 データ解析 |
生物種対応(ヒト・マウス・ラット以外) |
| 2群間比較解析の組み合わせ数の追加 | |
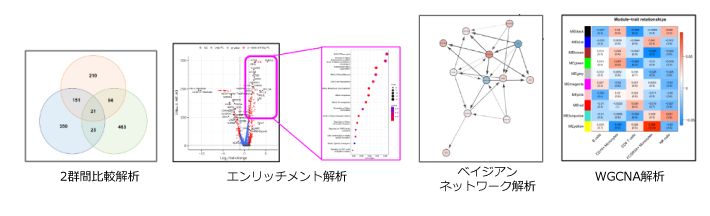
| 深堀解析:ベン図作成、追加エンリッチメント解析 | |
| WGCNA解析(既存DBに依存しない意味推定) | |
| ベイジアンネットワーク解析 (因果関係の推定) |
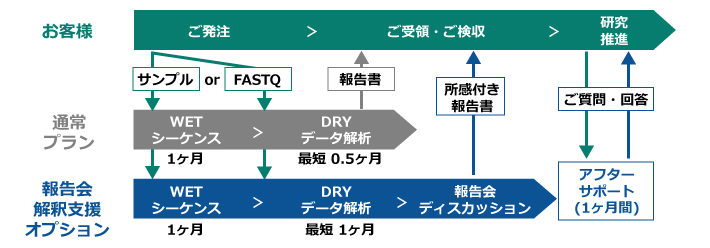
ご利用の流れ
RNA-seq解析とは
RNAseq解析とは、次世代シーケンサー(NGS)を用いて転写物の塩基配列を決定する方法です。この配列をリファレンスゲノムへアライメントし、転写産物毎の発現量を測定し、通常はサンプル間の発現量の比較解析を行います。スプライスバリアント毎の発現解析、融合遺伝子検出、変異解析、新たな転写産物の予測を行うことができます。
リファレンスゲノムのない生物の場合でも、配列をアセンブルして転写産物モデルを構築し、アライメントさせて解析することができます。
実験のポイント
解析対象のRNAの種類に適したライブラリー調整キットを用いて、ライブラリー作成を行います。必要なシーケンス量は、生物種や何をプロファイリングしたいかによって異なります。ヒトやマウスの場合、最低でも2000万リード、4Gbp以上の測定が推奨されます。スプライスバリアント(アイソフォーム)の検出や新規転写産物を探索する場合、もしくはFFPEサンプルやnon-conding RNAを扱う場合には、シーケンス量を増やす必要があります。
解析のポイント
NGSによるシーケンスで得られた配列は、FASTQというファイル形式で保存されます。この配列をスプライシングを考慮してゲノム配列にマッピングします。この結果は、BAM形式ファイルで保存され、IGV (Integrative Genomics Viewer)などのゲノムビューワーを用いて閲覧できます。
発現量は、リードカウント、または、補正値を用います。FPKM (Fragments Per Kilobase of exon per Million mapped fragments)やTPM (Transcripts per million)は、発現量をエクソン長と全マッピング数で補正した値です。CPM (Counts per Million mapped reads)は、発現量を全マッピング数で補正した値です。
2群間の比較解析では、発現量の比をlogスケールで表現した値、もしくはt検定による有意差P値がよく用いられます。
研究目的別高次解析のポイント
臨床上の特徴や薬剤が遺伝子発現にどのような影響を及ぼすのかを、発現変動が観測された遺伝子に注目し、特定のパスウェイや生物学的な機能のまとまりで、その意味を捉えることが高次解析の目的です。
このためには以下に示しますように、データの特徴を把握した上で2群間比較による発現変動遺伝子解析を行い、その上で研究目的に応じた生物学的な機能を理解するための解析手法を適切に選択することが重要となります。
データの特徴の把握
・主成分分析
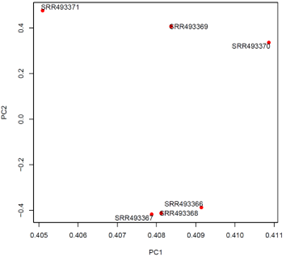
| 全サンプルを対象に、遺伝子発現の傾向を二次元プロットで表現します。これにより、二次元上にプロットされた各サンプル間の距離から、類似性を読み取ることができます。複数サンプルの発現プロファイルの類似度を視覚化し、その後の解析で群間の差を見出すことができるデータであることを確認します。 | |
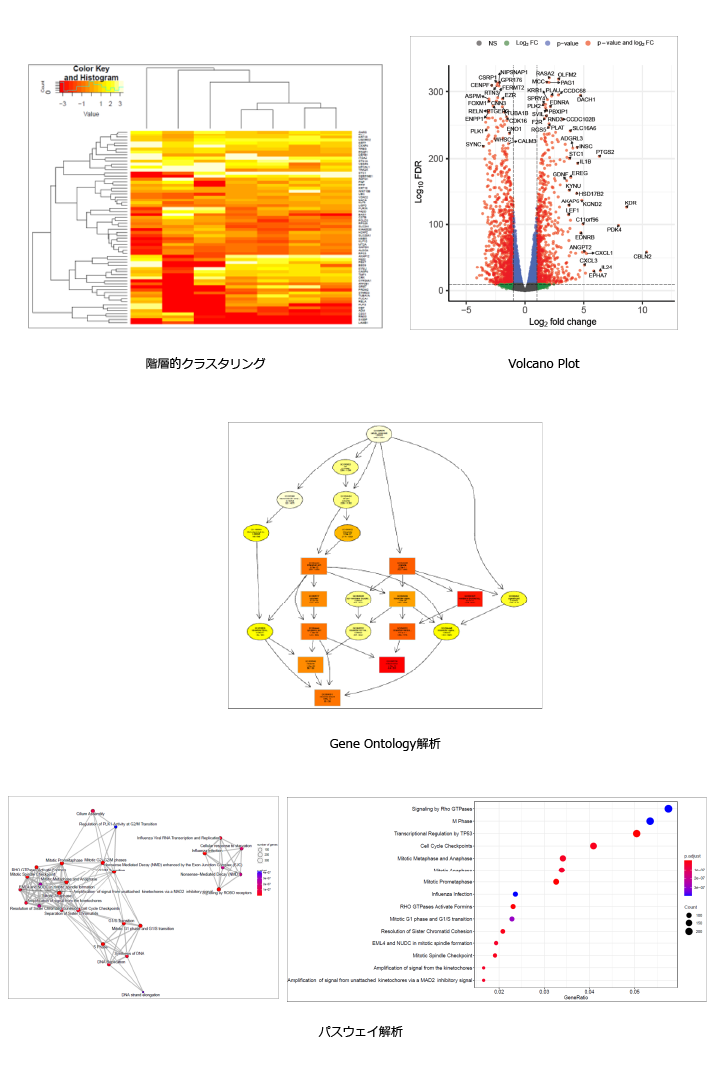
・階層的クラスタリングのヒートマップ

| 縦軸に遺伝子、横軸にサンプルを配置し、発現量を色で表現します。 各軸でクラスタリングを実施し、群ごとに同じまたは近いクラスタに分類します。はずれ値を持つサンプルを確認できます。 |
|
2群間比較による発現変動遺伝子解析
2群間で有意に発現量に差がある遺伝子群を特定します。
・Volcano plot
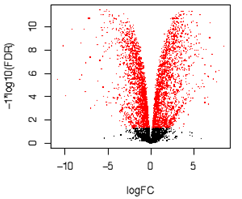
| 2群間の比較解析において、有意差P値(logスケール)と発現量の比(logスケール)を2次元プロットで表現します。各プロットの点は遺伝子に該当し、有意に変動している(p値や発現量比)遺伝子数の概観の確認や、変動の傾向の把握に使用します。 各点に該当する遺伝子名を重ねて表記するなどし、既知の遺伝子で想定される発現変動が起きているかを確認するなど、データの妥当性の検討に用います。 |
|
・Smear plot
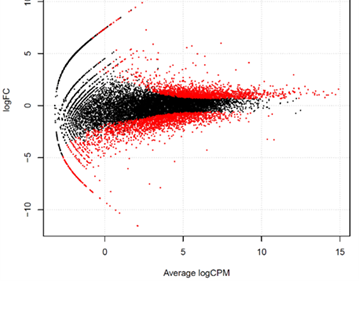
| 2群間の比較解析において、発現量の比(logスケール)と平均発現量(logスケール)を二次元プロットで表現します。これにより、十分な発現量が観測できている遺伝子が存在するか否か、また、その遺伝子数が多いか否かを確認します。発現変動比が大きい場合、少ない観測データに基づいた結果ではないことを確認し、判断が妥当かを確認することができます。 | |
発現変動遺伝子と生物学的意義
発現変動した遺伝子群の生物学的機能の共通点や、影響を受けているパスウェイ等の探索を行います
・GO(Gene Ontology)解析
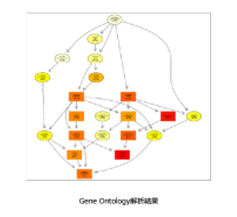
| Gene Ontologyとは、遺伝子の属性を説明する語彙を体系化・構造化した記述方法です。GOは生物学的プロセス、細胞の構成要素、分子機能の3カテゴリーに分けることができ、それぞれのカテゴリーにはさらなる下位概念が定義されています。有意に変動した遺伝子群がどのような生物学的概念にエンリッチしているかを解析することで、比較対象が影響を与えている生物学的機能を捉えます。 最上位GOから、下位GOまでの繋がりを図に示します。それぞれのGOのエンリッチメント解析結果は色の濃淡で有意差(P値)の大きさを表します。 |
|
・Pathway Enrichment解析
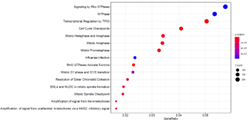
| 2群間の比較解析において、有意となった変動遺伝子群における、生物学的機能の推定を行います。 Reactomeなどのパスウェイデータベースに登録されている既知パスウェイに属する遺伝子セットを用い、有意に変動した遺伝子群がにエンリッチされたパスウェイを特定します。 横軸はパスウェイに属する遺伝子全体のうち有意に変動していた遺伝子の割合、縦軸は各パスウェイを示します。プロットのドットの大きさは有意に変動した遺伝子の数、色の濃淡は有意差(P値)の大きさを表します。 |
|
・GSEA (Gene Set Enrichment Analysis) 解析
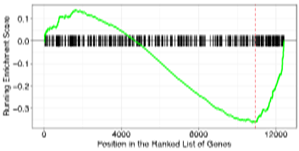
| 発現変動を調べた遺伝子群について、過去の研究結果から共通して関連があるとされている遺伝子セットへの偏りを推定します。遺伝子セットは既知のパスウェイデータベース等を参照し作成されています。 | |
アメリエフが選ばれる理由
- ・実験と解析をワンパッケージでご提供します
- ・研究目的にあった解析手法を選択し解析プランを組み立てます
- ・解析内容と解析結果を丁寧にご説明し仮説検証に貢献します
安心してご利用を開始していただけるよう、各担当スタッフがサポートします。

NGS
- RNA-seq解析
- シングルセルRNAseq解析
- シングルセル遺伝子発現Flex
- 空間的遺伝子発現解析
- 空間的遺伝子発現解析(Trekker™)
- WES遺伝性変異解析
- WESがん変異解析
- エピゲノム解析
アレイ
NGSロングリード
遺伝性疾患の研究
がん研究
精神疾患の研究
分子生物学研究