
2023.03.31
タグ別アーカイブ: 新年度応援キャンペーン|受託解析
2023.03.31
RNA-seq Wet&Dry解析【新年度応援キャンペーン】

論文作成には欠かせない
データ解析まで込みの
スペシャルプライス
3.9万円/サンプル
(税込42,900円)
※6サンプル以上の場合
サービス内容
【RNAシーケンス】| ○ ライブラリ調製 |
| ○ Illumina社 NovaSeq6000による(PolyA 4Gb/13.3Mペアエンドリード、non stranded RNA-seq)シーケンシング |
| ※4Gbを超える場合や、strand specific RNA-seqをご希望の場合はお問合せください |
| ○ 解析レポート、最終報告(手法解説、結果解釈のディスカッション) |
| ○ 発現定量解析:マッピング、 発現定量、階層的クラスタリング (ヒートマップ描画)、 主成分分析 |
| ○ 二群間の発現量比較解析:Volcano plot |
| ※サンプル数によって対応可能な組み合わせ数が異なりますのでご相談ください |
| ○ エンリッチメント解析:Gene Ontology解析、パスウェイ解析 (Reactome) |
サンプル要件 / 注意事項
サンプルタイプ:total RNA
| 総量(ng) | 液量(µL) | 濃度(ng/µL) | RIN値 | 純度 |
|---|---|---|---|---|
| 200以上 | 10以上 | 20以上 | 4.0以上 with flat base line |
OD260/280 = 1.8-2.2 OD260/230 ≥ 1.8 no degradation, no contamination |
- ※ サンプルQCでRNA総量またはRIN値が非常に低い場合は、再提出をお願いすることがございます。
- ※ シーケンシングはシンガポールにて実施します。
- ※ 海外輸送費・ 納品にかかる費用 (クラウドまたはHDD) を別途頂戴します。
- ※ ヒトおよびマウス、ラット以外の生物種をご希望の場合は、お問合せください。
ご利用の流れ

- 研究目的、サンプル情報をヒアリングし、お見積りを作成します。
- ご発注後、サンプルを準備いただき、シーケンスを行います。
- 報告会にて、高次解析結果の報告および解釈について、ご報告します。
RNAseq解析とは
RNAseq解析とは、次世代シーケンサー(NGS)を用いて転写物の塩基配列を決定する方法です。この配列をリファレンスゲノムへアライメントし、転写産物毎の発現量を測定し、通常はサンプル間の発現量の比較解析を行います。スプライスバリアント毎の発現解析、融合遺伝子検出、変異解析、新たな転写産物の予測を行うことができます。
リファレンスゲノムのない生物の場合でも、配列をアセンブルして転写産物モデルを構築し、アライメントさせて解析することができます。
実験のポイント
解析対象のRNAの種類に適したライブラリー調整キットを用いて、ライブラリー作成を行います。必要なシーケンス量は、生物種や何をプロファイリングしたいかによって異なります。ヒトやマウスの場合、最低でも2000万リード、4Gbp以上の測定が推奨されます。スプライスバリアント(アイソフォーム)の検出や新規転写産物を探索する場合、もしくはFFPEサンプルやnon-conding RNAを扱う場合には、シーケンス量を増やす必要があります。
解析のポイント
NGSによるシーケンスで得られた配列は、FASTQというファイル形式で保存されます。この配列をスプライシングを考慮してゲノム配列にマッピングします。この結果は、BAM形式ファイルで保存され、IGV (Integrative Genomics Viewer)などのゲノムビューワーを用いて閲覧できます。
発現量は、リードカウント、または、補正値を用います。FPKM (Fragments Per Kilobase of exon per Million mapped fragments)やTPM (Transcripts per million)は、発現量をエクソン長と全マッピング数で補正した値です。CPM (Counts per Million mapped reads)は、発現量を全マッピング数で補正した値です。
2群間の比較解析では、発現量の比をlogスケールで表現した値、もしくはt検定による有意差P値がよく用いられます。
研究目的別高次解析のポイント
臨床上の特徴や薬剤が遺伝子発現にどのような影響を及ぼすのかを、発現変動が観測された遺伝子に注目し、特定のパスウェイや生物学的な機能のまとまりで、その意味を捉えることが高次解析の目的です。
このためには以下に示しますように、データの特徴を把握した上で2群間比較による発現変動遺伝子解析を行い、その上で研究目的に応じた生物学的な機能を理解するための解析手法を適切に選択することが重要となります。
データの特徴の把握
・主成分分析
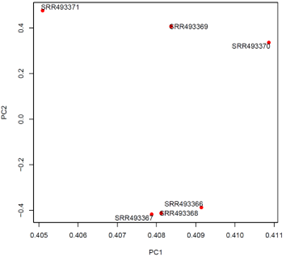
| 全サンプルを対象に、遺伝子発現の傾向を二次元プロットで表現します。これにより、二次元上にプロットされた各サンプル間の距離から、類似性を読み取ることができます。複数サンプルの発現プロファイルの類似度を視覚化し、その後の解析で群間の差を見出すことができるデータであることを確認します。 | |
・階層的クラスタリングのヒートマップ

|
縦軸に遺伝子、横軸にサンプルを配置し、発現量を色で表現します。 各軸でクラスタリングを実施し、群ごとに同じまたは近いクラスタに分類します。はずれ値を持つサンプルを確認できます。 |
|
2群間比較による発現変動遺伝子解析
2群間で有意に発現量に差がある遺伝子群を特定します。
・Volcano plot
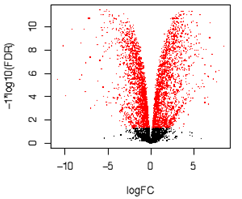
|
2群間の比較解析において、有意差P値(logスケール)と発現量の比(logスケール)を2次元プロットで表現します。各プロットの点は遺伝子に該当し、有意に変動している(p値や発現量比)遺伝子数の概観の確認や、変動の傾向の把握に使用します。 各点に該当する遺伝子名を重ねて表記するなどし、既知の遺伝子で想定される発現変動が起きているかを確認するなど、データの妥当性の検討に用います。 |
|
発現変動遺伝子と生物学的意義
発現変動した遺伝子群の生物学的機能の共通点や、影響を受けているパスウェイ等の探索を行います
・GO(Gene Ontology)解析
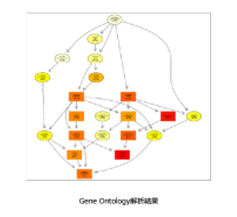
|
Gene Ontologyとは、遺伝子の属性を説明する語彙を体系化・構造化した記述方法です。GOは生物学的プロセス、細胞の構成要素、分子機能の3カテゴリーに分けることができ、それぞれのカテゴリーにはさらなる下位概念が定義されています。有意に変動した遺伝子群がどのような生物学的概念にエンリッチしているかを解析することで、比較対象が影響を与えている生物学的機能を捉えます。 最上位GOから、下位GOまでの繋がりを図に示します。それぞれのGOのエンリッチメント解析結果は色の濃淡で有意差(P値)の大きさを表します。 |
|
2023.03.31
シングルセルRNA-seq Wet&Dry解析【新年度応援キャンペーン】

論文作成には欠かせない
データ解析まで込みの
スペシャルプライス
103万円/サンプル
(税込1,133,000円)
サービス内容
【シングルセルRNAシーケンス】| ○ 10x Genomics社 Chromiumによるライブラリー調製 |
| ○ Illumina社 NovaSeq6000による120Gbのシーケンシング |
| ○ Cell Rangerによる発現定量 |
| ○ 解析レポート、中間報告および最終報告(手法解説、結果解釈のディスカッション) | ||
| ○ 研究の目的に応じて下記よりご提案 | ||
| ● 発現変動遺伝子の群間比較解析 | ||
| 細胞クラスターごとの特徴遺伝子検討 サンプル間発現比較解析 GO解析(Gene Ontology) パスウェイ解析 |
||
| ● 細胞分化に関わる発現変動遺伝子解析 | ||
| 疑似系譜解析(Cell trajectoryの推定) | ||
サンプル要件 / 注意事項
サンプルタイプ:細胞
- 凍結状態であること。
- 細胞数は106細胞以上であること。
- 融解後の細胞の生存率が90%以上推奨。
- 細胞は分散もしくは単離してください。
- EDTA0.1mM以下、マグネシウム3mM以下、界面活性剤は含まないこと。
- 細胞サイズが直径30μm以下であること。
- 40μmのフィルターにかけてください。
- 凍結の際にはセルバンカー等を用いてください。
- ※ ヒトおよびマウス以外の生物種をご希望の場合は、お問合せください。
- ※ 納品にかかる費用 (クラウドまたはHDD) を別途頂戴します。
- ※ シングルセル遺伝子発現解析は、機器を持ち込んでの出張サービスも承っております。その際には出張費をご請求いたします。また、サンプルを持ち込んで頂いてのご利用も可能です。詳細はお問合せください。
ご利用の流れ

- 研究目的、サンプル情報をヒアリングし、お見積りを作成します。
- ご発注後、サンプルを準備いただき、シーケンスを行います。
- 中間報告会にて、途中結果の報告および解析方針の確認、内容についてのディスカッションを行います。
- 最終報告会にて、結果の報告および内容について、ご報告します。
シングルセルRNAseq解析とは
シングルセルRNAseqとは、細胞の遺伝子発現プロファイルを一細胞レベルで取得して観測する画期的な技術です。これまでのRNAseq解析では、観測対象が細胞集団であり、細胞集団における各遺伝子の"平均的な"遺伝子発現を測定していました。一方、scRNAseqは細胞集団内の個々のトランスクリプトームを捉え、細胞特異的な変化を同定することが可能になります。Science誌において2018年のBreakthrough of the Yearとしても選出された技術であり、近年盛んに研究に用いられています。
実験のポイント
シングルセルRNAseqでは、細胞の生存率が非常に重要です。
死細胞が溶解すると周囲にRNAが遊離し、分析時のバックグラウンドノイズの一因となり、シングルセルデータの品質を著しく低下させるため、細胞の生存率を高める必要があります。一般的には生存率90%以上が推奨され、クリアな解析結果を得るためには、80%程度以上の生存率が必要です。
またライブラリ調整に際しては、細胞数が106以上であること、細胞サイズが直径 30μm以下であること、細胞を分離させておくことなどの条件をクリアする必要があります。
解析のポイント
特定の細胞種ごとの遺伝子発現プロファイルを確認します。まず、データのクオリティを確認した上で、細胞をクラスタリングし、各クラスターごとに遺伝子発現の状態を把握するための一連の解析を行います。
高次解析により得られる図表の例
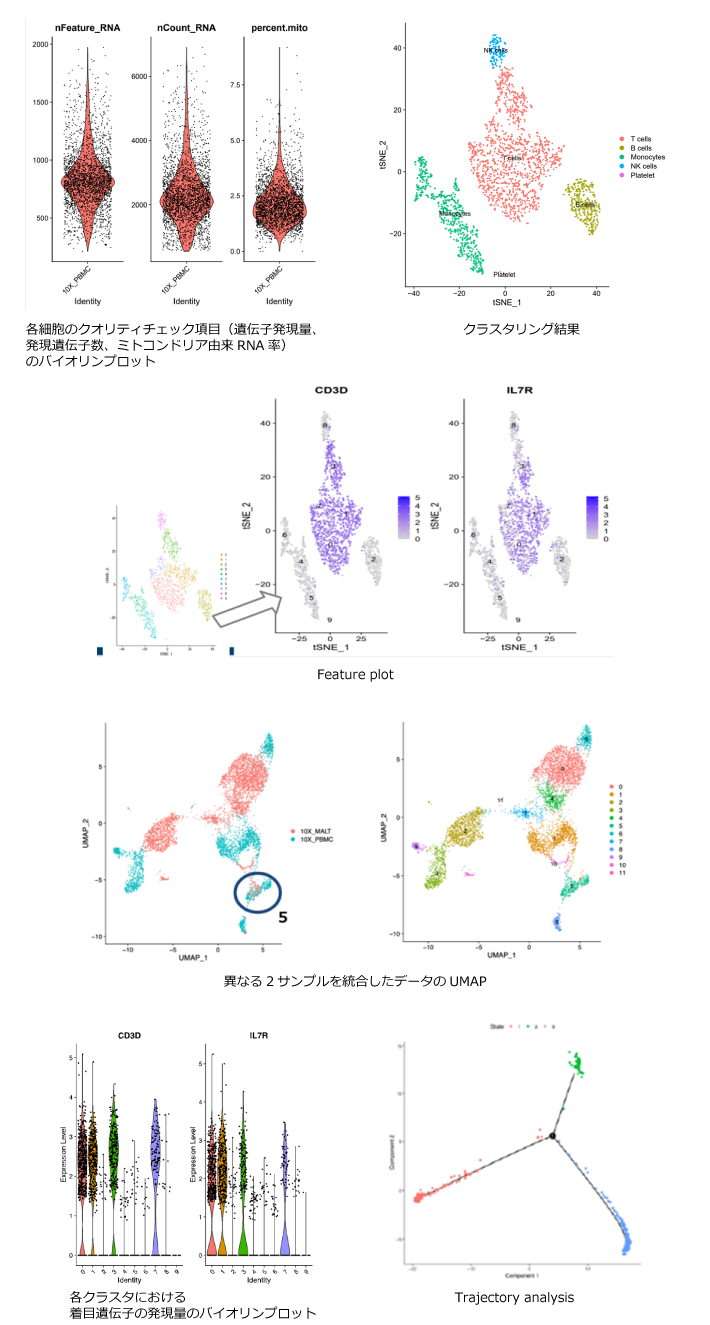
研究事例
1)がん組織の不均一性における薬物耐性の発生機序解明に関する研究
- 薬物耐性獲得のメカニズムの解明は、がんの薬物治療において不可欠な要素です。耐性獲得の背景は、様々あるとされていますが、がん組織の不均一性がその原因の一つであると考えられています。Kashimaらは、がん組織の不均一性における薬物耐性の発生機序解明にscRNAseq解析を利用しました(Kashima et al., Cancer Res, 2021)。
- 具体的には、オシメルチニブ耐性細胞の出現機構を解明するために、EGFR遺伝子に変異をもつヒト肺腺がん細胞株であるNCI-H1975細胞を用いて一細胞解析を行いました。H1975はオシメルチニブ感受性の細胞であることが知られていますが、この細胞を段階的にオシメルチニブに曝露し、各段階での細胞群をscRNAseq解析することで、①親株に少数のオシメルチニブ耐性細胞が存在すること、②オシメルチニブ曝露群にしか出現しない細胞群が存在すること、が明らかになりました。
2)発生/分化分野での毛包形成研究
- 全世界で男性の50%、女性の25%が薄毛に悩んでいると言われており、自己由来の幹細胞から毛包を誘導する研究が注目されています。しかし、生体内での毛包のde novo形態形成については、毛包の不均一性や非同期的な発達のため、アプローチが難しく理解が進まない状態でした。このような観点から、毛包のde novo形態形成の基礎となる分子経路を明らかにすることは、毛包の発生に関する深い洞察をもたらし、in vitro条件下での毛包の発生を誘導する上で重要な意味を持つと考えられます。
- GeらはscRNAseq技術を用いて、様々な分化期におけるマウス背部皮膚から得られた細胞群に対して一細胞のトランスクリプトームを統合的に解析しました(Ge et al., Theranostics, 2020)。その結果、9つの主要な細胞集団の同定と、上皮/真皮細胞系の分化の軌跡を構築することに成功し、細胞の運命決定に関わる主要なレギュロンが順次活性化されていることを明らかにしました。
- </li >