
Xenium(空間的遺伝子発現解析)|データ解析【受託解析キャンペーン】

論文作成には欠かせない
パスウェイ・GO解析
擬似系譜解析
までをプロが支援
43万円/サンプル(税抜)
※1~3サンプルの場合
※他のプラットフォーム(Visium HD、Stereo-seq™
CosMx™等)もWETから対応可能です。
▼データ解析の専門家による報告会の様子をご覧ください▼
データ解析内容
【発現定量解析】| ○ クオリティコントロール |
| ○ 正規化、データ統合 |
| ○ 次元削減 |
| ○ クラスタリングおよび UMAP上の可視化・空間座標上の可視化 |
| ○ クラスタごとの特徴遺伝子の探索 |
| ○ 着目遺伝子の発現量可視化(Feature plot, Violin plot) |
| ○ 各クラスタおけるサンプル間発現比較解析 |
| ○ Gene Ontology解析、Reactomeパスウェイ解析 |
| ○ Cell trajectoryの推定 |
| ○ Pseudotimeの推定 |
| ○ 着目遺伝子の発現パターン可視化 |
受入れデータ・納品物
受入れデータ形式
- 発現マトリクス
※ファイル形式を予めご教示ください - 組織切片画像およびイメージ処理済みデータ
- 組織切片上の位置情報を含むデータ
納品物
- 解析結果の中間報告資料(PDF)
- 解析結果の最終レポート(PDF)
- データ解析結果(PDF、Excel)
解析結果レポート例
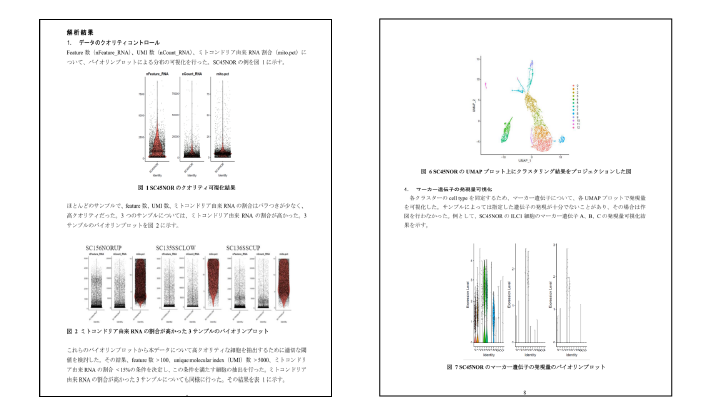
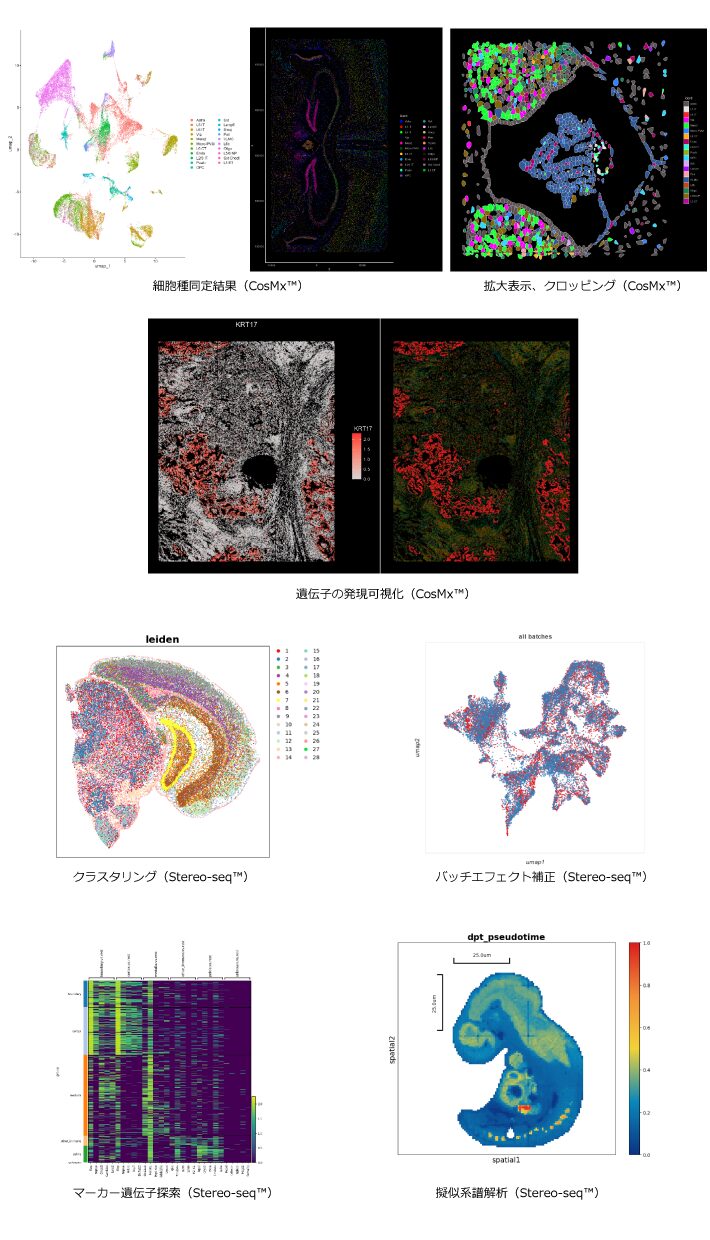
バイオマーカー探索を目的としたデータ解析を実施した場合の報告書例です。研究目的に応じたご報告を行います。
価格表(税抜)
| 製品名 | 単価 | |
|---|---|---|
| データ解析 (Xenium、Visium HD、Stereo-seq™、CosMx™) |
1~3サンプル | 43万円/サンプル |
| 4サンプル以上 | 都度見積 | |
- ※4サンプル以上はボリュームディスカウントが適用されますのでご相談ください。
- ※1サンプル=1切片という考え方になります。1スライド上に多数の切片を乗せる場合があるため、ボリュームディスカウントは都度見積となります。
空間的遺伝子発現解析とは
空間的遺伝子発現解析は、生物組織内の特定の部位における遺伝子発現の可視化を行う技術です。この技術は、2020年には約500件の研究論文が発表されていましたが、2023年にはその数が倍増し、今後もますます研究と応用の幅が広がっていくことが期待されています。
この技術の特徴は、組織内の遺伝子発現を空間的に捉え、各スポットの遺伝子発現プロファイルを解析する点にあります。例えば、スポット間の遺伝子発現の差や、細胞間の相互作用を解析するために活用されています。従来のバルクRNA発現解析では、組織全体の平均的な遺伝子発現しか把握できませんでしたが、空間的遺伝子発現解析により、より高解像度な細胞レベルでの解析が可能となりました。
技術の進化と利点
従来のバルクRNA発現解析では、組織全体の遺伝子発現量を一括で解析するため、平均化されたデータしか得られませんでした。これに対して、シングルセルRNA解析では、個々の細胞の遺伝子発現パターンを把握することができ、細胞間の違いや介入前後の変化も詳細に解析できるようになりました。このシングルセルRNA解析の発展が、空間的遺伝子発現解析の登場へと繋がっています。
さらに、空間的遺伝子発現解析では、2次元あるいは3次元情報を利用して組織内の遺伝子発現を解析します。これにより、組織内の微少環境解析や細胞間の相互作用を詳細に観察することが可能です。
代表的なプラットフォームとその特徴
現在、空間的遺伝子発現解析のプラットフォームとして、CosMx™(NanoString Technologies社)、Visium HD・Xenium(10x Genomics社)、Stereo-seq™(MGI社)などが主に利用されています。これらのプラットフォームは、解析に使用する遺伝子数や解像度が異なるため、目的に応じた選択が求められます。
解析のポイント
空間的遺伝子発現解析では、空間自己相関やホットスポット解析など、空間情報を考慮した解析手法を用いることで、遺伝子発現を組織画像上にマッピングするなど、空間情報を視覚的に表現することで、データの解釈が容易になります。
組織の不均一性や実験操作によるノイズは、データ解析の精度を低下させる可能性があります。具体的には、ライブラリーサイズや遺伝子長などの影響を補正するために、適切な正規化方法を選択する必要があります。また、実験日や操作者などの違いによるバッチ効果を補正することで、データの信頼性を向上させることができます。
細胞の種類を自動的に分類する手法もありますが、解釈には注意が必要です。既知のマーカー遺伝子の発現パターンと比較することで、細胞タイプの解釈の精度を高めることができます。また、シングルセルRNA-seqなど他のデータと統合することで、細胞タイプの解釈をより確実にすることができます。